弓 亜玲
(ビス(N,N-ジメチルカルバモイルカルコゲノ)メタン類と四塩化スズの反応による カルコゲノアルデヒドの新規な合成)
Recently, various methodologies for the synthesis of chalcogenoaldehydes have been reported in the light of their structural interests, their potentialities as new reactive intermediates for the organic reactions, and the increasing interests in the biological activities for some organoselenium compounds. However, most methods required multi-step processes to prepare the suitable precursors for chalcogenoaldehydes. In the course of our studies on the novel generation of highly-reactive species related to higher-row chalcogenocarbonyl compounds, we found a convenient preparation of stable diseleno- and ditelluroacetals derivatives using the reaction of N,N-dimethylchalcogenocarbamate ions with gem-dihaloalkanes. Then I carried out the further reaction of unsymmetrical C-Se or C-Te bond cleavage of this symmetrical diseleno- or ditelluroacetals to generate selenoaldehydes or telluroaldehydes.
Bis(N,N-dimethylcarbamoylchalcogeno)methanes 3 were efficiently prepared through a stepwise treatment of diselenide or ditelluride 1 with hydride NaH or NaBH4, then a gem-dihaloalkane 2 under an Ar atmosphere.
When bis(N,N-dimethylcarbamoylseleno)methanes 3(X=Se) were treated with SnCl4 at R.T. under an Ar atmosphere, β-1,3,5-triselenanes 5 (X=Se), the trimers of selenoaldehyde 4 (X=Se), were obtained as the main product. With the trapping experiments by using allyltrimethylsilane or 2,3-dimethyl-1,3-butadiene, the successful separation of corresponding allylation products 6 (X=Se) or cycloadducts 7 (X=Se) just suggested that the conversion of bis(N,N-dimethylcarbamoylseleno)methanes 3 into selenoaldehydes 4 involving the formation of acylselonium ion A through weak coordinating interaction of SnCl4 with 3 at low temperature and the subsequent removal of N,N-dimethylcarbamoyl cation B from A. β-1,3,5-Triselenanes 5 were assumed to be afforded through elimination of stable N,N-dimethylcarbamoyl cation B from A and the subsequent trimerization of the resulting selenoaldehydes 4 in the final stage.
Treatment of bis(N,N-dimethylcarbamoyltelluro)methanes 3 (X=Te) with SnCl4 also afforded the 1,3,5-tritelluranes 5 (X=Te), the trimers of telluroaldehyde 4 (X=Te). 1H NMR monitoring experiment of bis(N,N-dimethylcarbamoyl telluro)phenylmethane with SnCl4 in CDCl3 at 25 ℃ just gave the information that 2,4,6-triphenyl-1,3,5-tritellurane was formed as the main product along with the formation of benzaldehyde 8a (X=Te, R=C6H5) and ditelluride 1 (X=Te). With the standing time, the increasing of benzaldehyde 8a and the decreasing of 1,3,5-tritellurane 5 suggested a transformation from tellurobenzaldehyde 4a (X=Te, R=C6H5) to benzaldehyde 8a. Trapping experiments were carried out by using allyltrimethylsilane or 2,3-dimethyl-1,3-butadiene to afford the corresponding allylation products or [4+2] cycloadducts.
All these results are shown in Scheme 1.
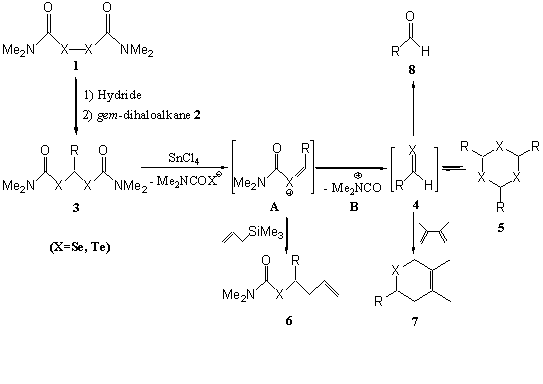
Scheme 1
In conclusion, selenoaldehydes were successfully generated by the reaction of bis(N,N-dimethylcarbamoylseleno)methanes with SnCl4 through a stepwise fragmentation pathway involving the formation of acylselonium ion. And telluroaldheydes were also generated by the similar reaction of bis(N,N-dimethylcarbamoyltelluro)methanes with SnCl4. Further research work on the seperation and isolation of 1,3,5-tritellurane, cycloadducts 6 (X=Te) and allylation products 7 (X=Te) are under way.